
www.bcb.lon.ac.uk
Summary
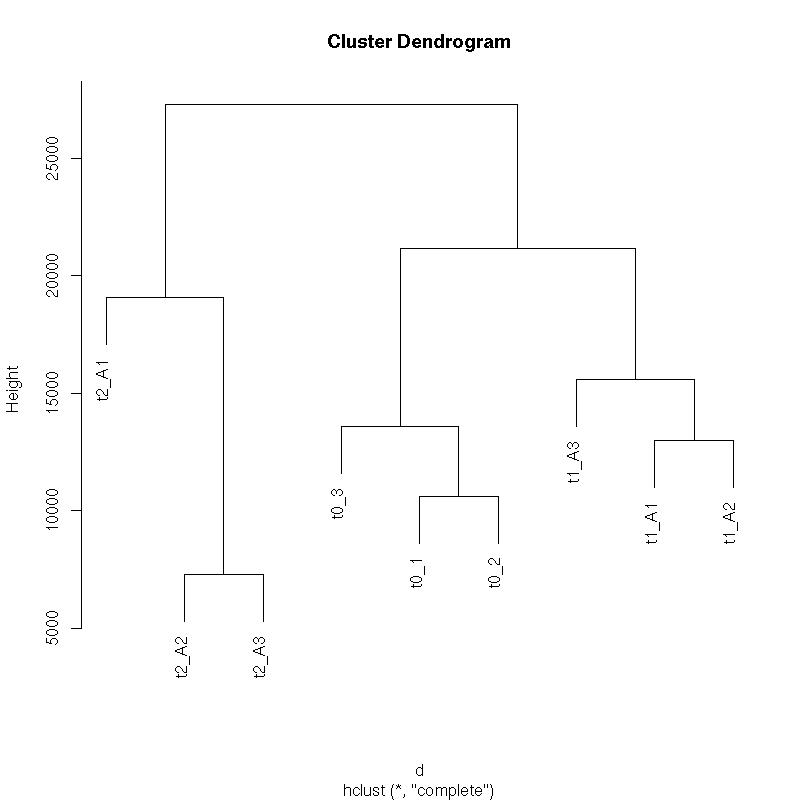
Sample Clustering
Differential Expression using Limma
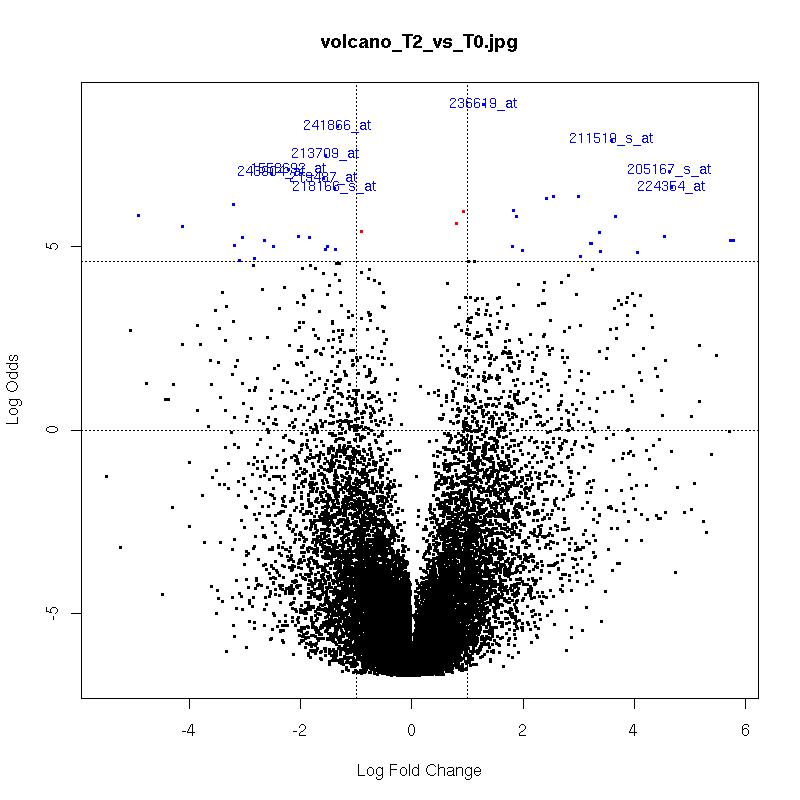
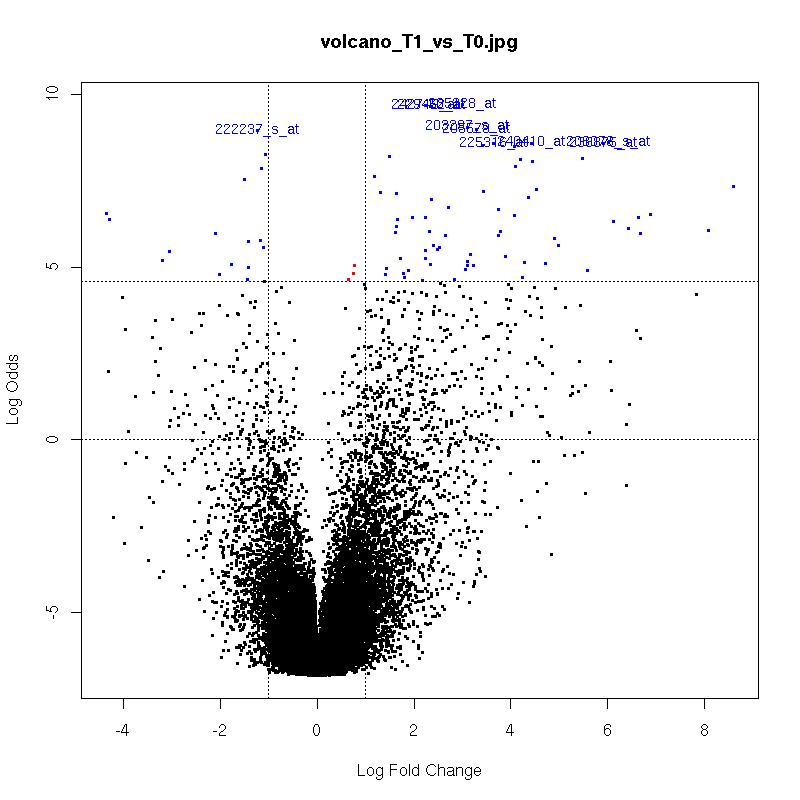
Volcano Plots
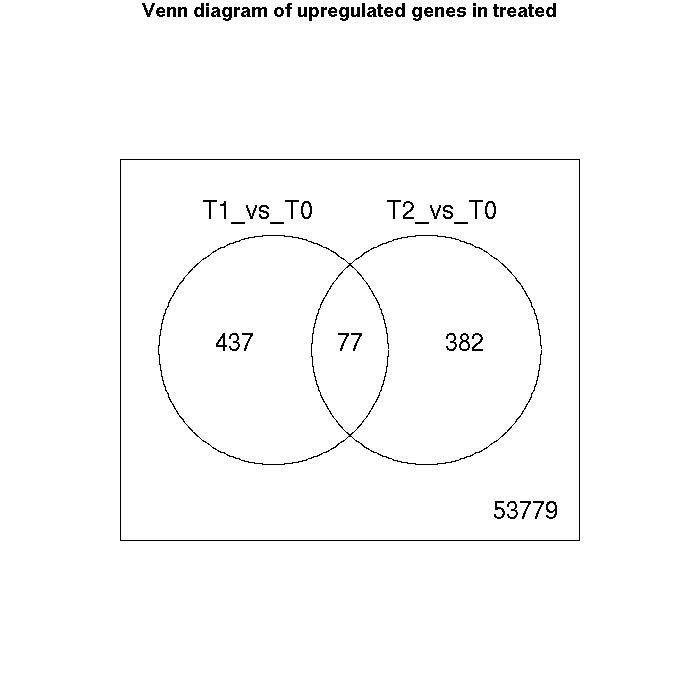
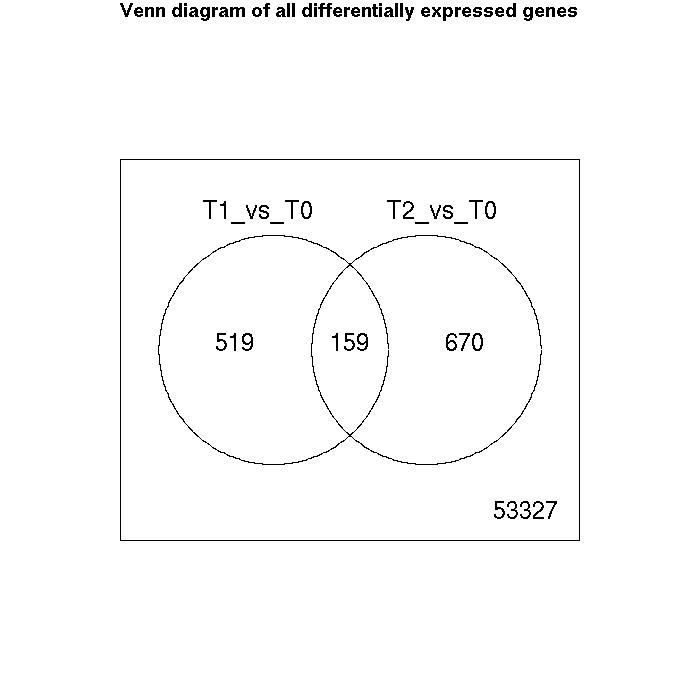
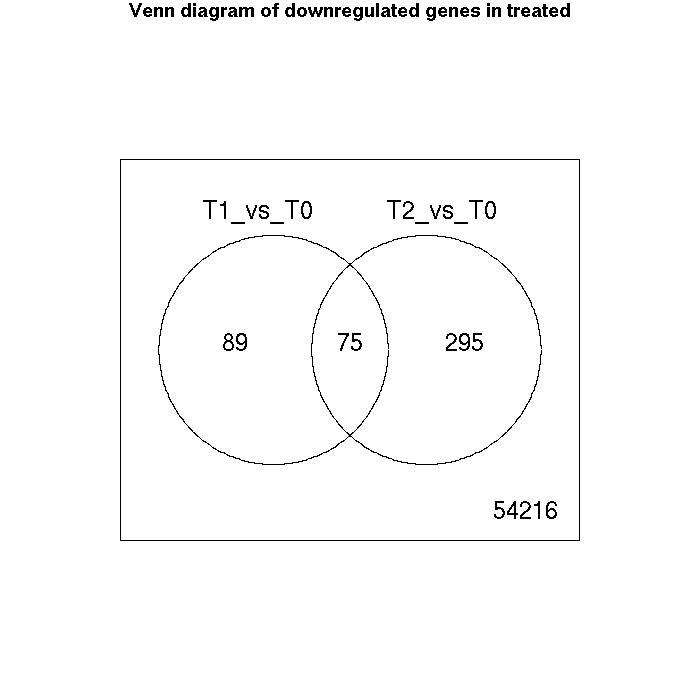
Venn Diagrams
Appendix
| Customer: | Joe Blogg | |
| Author: | Sonia Shah | |
| Chip Type: | Human xyz | |
| Summary: |
| array index | file | name | condition |
| 1 | t0_1.CEL | t0_1 | untreated_T0 |
| 2 | t0_2.CEL | t0_2 | untreated_T0 |
| 3 | t0_3.CEL | t0_3 | untreated_T0 |
| 4 | t1_A1.CEL | t1_A1 | treated_T1 |
| 5 | t1_A2.CEL | t1_A2 | treated_T1 |
| 6 | t1_A3.CEL | t1_A3 | treated_T1 |
| 7 | t2_A1.CEL | t2_A1 | treated_T2 |
| 8 | t2_A2.CEL | t2_A2 | treated_T2 |
| 9 | t2_A3.CEL | t2_A3 | treated_T2 |
 |
Comment:
All biological replicates cluster together. As expected, the first time point T1 has expression that is more similar to time 0 (T0) than the T2 later time point
summary_TreatedvsUntreated.txt
| Number of genes meeting significance criterion Benjamini Hochberg (FDR) less than 0.05 | T1_vs_T0 | T2_vs_T0 |
| downregulated | 164 | 370 |
| no change | 53997 | 53846 |
| upregulated | 514 | 459 |
The lists of significant genes for each comparison have been saved as excel files.
Links to external databases for all the differentially expressed genes can be found in the T1_vs_T0.html and T2_vs_T0.html files.
plot of log-fold changes versus log-odds of differential expression.
The x-axis indicates the log2 value of fold-change between the two conditions.
Genes in the lower left and right squares of the graph are probably false positives.
Click on the images for a larger image
 |
 |
 |
 |
 |
The data was analysed using Bioconductor v1.5 and R version 2.1.0
Bioconductor: open software development for computational biology and bioinformatics
Wu et al., GCRMA normalisation
Miller et al. Simpleaffy package
Gentleman et al. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer, 2005
Linear Models (limma) Statistics
The Linear Models package uses Bayesian statistics to compute the probability of a gene being differentially expressed in any defined contrast. Bayesian statistics is a means of measuring the probability of an outcome (i.e. a gene being differentially expressed), calculated from a ratio of the probabilities of the experiment outcome and the prior assumption of the experiment outcome (the null hypothesis - nothing is differentially regulated).
The summary statistics are computed for each gene and each contrast (i.e. experiment comparison such as treated vs. control). These include:
M-value(M) - log2 fold change for that gene. A positive value indicates up-regulation of a gene, a negative value indicates down-regulation.
A-value(A) - average expression value for that gene across all the arrays
t - moderated t-statistic is the ratio of the M-value to its standard error. This has the same interpretation as the ordinary t.statistic except that the standard errors have been moderated across genes, effectively borrowing information from the ensemble of genes to aid with inference about each individual gene.
p-value - obtained from the distribution of the moderated t-statistic, usually after some form of adjustment for multiple testing such as Bonferroni or Benjamini-Hochberg.
B-statistic (B or lods or Log Odds) - is the log odds that the gene is differentially expressed. A B-statistic of zero corresponds to a 50-50 chance that the gene is differentially expressed. That is, a B-value of zero means the probability of the gene being differentially expressed (the outcome of the experiment) is equal to the probability that it is not differentially expressed (null hypothesis).
Example : A gene has a B-value of 1.5. The odds of
differential expression is exp(1.5)=4.48, or about 4.5 to 1. The
probability that the gene is differentially expressed is
4.48/(1+4.48)=0.82 or 82%
B-value = 0.3, odds = 1.35 to 1, probability = 57%
B-value = 4.6, odds = 99 to 1, probability = 99%
The B-value and the moderated t-statistic rank the genes in the same order (given that there are no missing values in the data). p-values and B-values also usually rank the genes in the same order. All three measures are closely related. A low p-value, and a high B-value should indicate the ranking (i.e. most significant=1 to least significant=54675 (or number of genes on array)) in the dataset for that particular contrast. The ranking will vary for each gene depending on the comparisons made between samples.
Multiple Correction
The most common form of multiple testing is "fdr" which is Benjamini & Hochberg's method to control the false discovery rate. If all genes that have an fdr-adjusted value of less than the threshold, let say 0.05, are considered as differentially expressed then the expected proportion of false discoveries is controlled to be less than the threshold value, in this case 5%.
In another method, the Bonferroni correction, the chance of making even a single type I error (false positive) can be maintained at the desired level, which in this case 5%. Bonferroni is more stringent than the Benjamini-Hochberg method.
Annotation of probesets
Probeset annotation is derived from the NetAffx site (www.affymetrix.com, free access but login required). Please note that while the NetAffx annotation is revised regularly we recommend that the annotation is confirmed before further experiments such as qPCR. This can be done by Blasting the probe or probe target sequence against the human genome. Alternatively the probe alignment can be checked in the EnsEMBL data or by querying the Adapt database .