Go to DomPred Background
Input sequence (single letter code)
DomPred lower sequence length limit
PSI-BLAST sequence alignment domain prediction
DomSSEA domain prediction
Include secondary structure profile plot
Attach results to email?
Format of Results
Input sequence (single letter code)
Type
or cut and paste your query sequence into the form (no nucleic acid
sequences please!). The sequence must be in the format of the amino
acid single letter code, in either capital and or lower-case letters.
Spaces within the pasted sequence are permitted - however note that
these will be parsed out prior to analysis.
We recommend that you enter your sequence as a plain single-letter string like this: ALGSNLNTPVEQLHAALKAISQLSNTHLVTTSSFYKSKPLGPQDQPDYVNAVAKIETELS
DomPred lower sequence length limit
Figure 1.
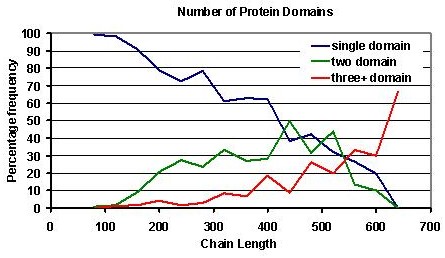
Of course it is possible for a chain of 120 residues or less to consist of two-domains, and if you have such suspicions about your query, it is worth considering that over 99% of domains in the CATH database are over 40 residues in length, it is therefore likely that the domain boundary in such a case will be located approximately midway between the amino and carboxyl termini of the query sequence.
PSI-BLAST sequence alignment domain prediction
The PSI-BLAST sequence alignment domain prediction searches the query sequence against a large database of sequences (nrdb90), including sequences from Pfam-A.
Pfam-A search
Domain sequences
from Pfam-A are searched against the query sequence, and if significant
sequence matches are found (as defined by the chosen E-value cut-off),
this is indicated on the DomPred results page. A separate table
displaying such hits accessible from the results page.
Homology to
largely complete Pfam-A sequences may be found over all or part of the
query sequence, thus indicating a putative single or multi-domain chain
respectively.
Query vs sequence database
In cases where
no clear homology exists to known domain sequences, such as Pfam-A
domain sequences, a different strategy is required. Here, the query
sequence is searched against a non-redundant sequence database
(nrdb90), utilising the given parameters specified in the input form to
identify significantly matching sequence homologues. These matching
sequences are then used to identify possible domain boundaries within
the query sequence (and therefore predict if single or multi-domain).
The domain
boundary prediction procedure utilises an algorithm to identify residue
positions to which the N and C termini of matching database hits are
aligned to the query sequence. The positions of the N and C termini
from all the PSI-BLAST database matches are simply summed along the
query sequence. Cases where both N and C termini hits are found in
similar regions along the query sequence are given a higher weighting.
The summed
profile is then smoothed using a window of 15 residues, and Z-score's
calculated over this profile. Significant peaks (Zscore>1.5) over
the mean termini value of the query are assigned as putative domain
boundaries. Termini hits to the first and last 50 residues of the
alignment profile are not considered as these regions often contain a
large multiple of alignment termini that correspond to the true termini
ends of the query sequence.
The alignment
profile generated by the PSI-BLAST alignments (and drawn by gnuplot) is
shown at the top of the results page. Putative domain boundaries are
indicated by peaks in the plot. Peaks considered to be significant by
the algorithm are indicated.
In cases where
significant peaks are found, and the query sequence is predicted to be
multi-domain, multi-domain predictions given by DomSSEA are given
higher significance.
Input E-value cut-off (default 0.01)
Optimisation of
the PSI-BLAST sequence alignment domain prediction showed an E-value
cut-off of 0.01 to give the best trade-off between the sensitivity and
selectivity (define?) of domain boundary prediction. Decreasing the
E-value (ie reducing the number of 'significant' aligned sequences) was
found to reduce sensitivity however increase the selectivity of domain
boundary prediction.
Input number of PSI-BLAST iterations (default 5)
The default
number of PSI-BLAST iterations used is 5. Decreasing the iteration
number may increase the speed of the PSI-BLAST search, but my also
result in the failure to identify more distant homologues. The user
should be aware that the higher the iteration value the higher the risk
of introducing profile wander into the PSI-BLAST sequence search.
DomSSEA domain prediction
In cases where no significant sequence matches have been found to Pfam-A sequences, or no significant domain termini peaks found by the PSI-BLAST alignment algorithm, DomSSEA can be used to predict the domain content of you query sequence.As outlined in Marsden et al. (In Press), DomSSEA is based on the idea that a crude fold recognition algorithm based on the mapping of predicted secondary structures to observed secondary structure patterns in domains of known 3-D structure might be reliable enough to parse a long target sequence into putative domains. This is often the way in which a human sequence analyst will attempt to parse a protein into domains when homology-based approaches have been unsuccessful.
Secondary structure element alignments methods (SSEA) have been shown to provide a rapid prediction of the fold for given sequences with no detectable homology to any known structure and have also been applied to the related problem of novel fold detection (McGuffin et al., 2001; McGuffin & Jones, 2002).
DomSSEA results table
An example of
the DomSSEA results table output is shown below. The table lists the
prediction results in decending order of the SSEA score. Information
about the aligned chain from the DomSSEA library can be accessed by
clicking on appropriate PDB code which takes you to its PDBSum
entry. The predicted number of domains and corresponding domain
boundaries are listed, as are the CATH 'CAT' codes of the domain(s)
aligned to the query sequence. A dash (-) is placed in the domain
boundary column for single domain predictions as single domains have no
inter-domain boundarys. A '?' is used in cases where a boundary
assignment is unclear.
![]()
Score
Match
SSEA
No. Doms
Boundaries
CATH code
![]()
0.831
7mdhB
Show SSEA
2
187
3 40 50
, 3 90 110
![]()
0.823
1mldD
Show SSEA
2
172
3 40 50
, 3 90 110
Include secondary structure profile plot
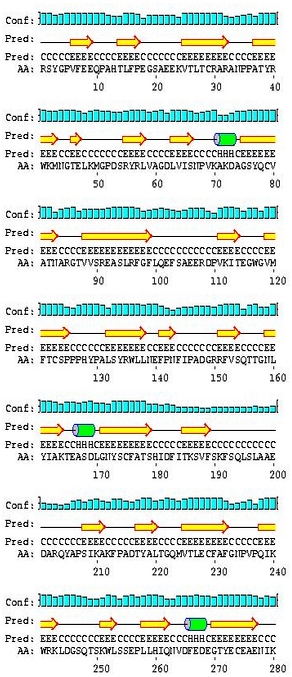
Multi-domain proteins may contain regions of different secondary structure class. For example a two-domain chain may contain an all-beta domain, followed by an all-alpha domain. The transition between such regions may be enough to predict a putative domain boundary.Secondary structure predictions are made for query sequences by PSIPRED. Using a smoothing window of 9 residues, a profile of the secondary structure can be shown on the plot shown at the top of the results page figure (2).
Show PSIPRED prediction
The graphical
output generated by PSIPRED can be shown on the results page if
specified. .Pdf and .PS versions of these predictions can be
downloaded.For more information on PSIPRED go to the PSIPRED homepage www.PSIPRED.net.
Attach results to email?
All results data from the results form can be emailed. Note however this may result in a large amount of data being sent to your email server.Format of Results
The response email provides a link to a web page containing the DomPred results for the protein submitted. For example, if we submit the AXONIN-1 protein (PDB ID 1CS6) to the DomPred server, the following result page is generated:
 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 contact contact |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Marsden,R.L., McGuffin,L.J. & Jones,D.T. 2002. Rapid protein domain assignment from amino acid sequence using predicted secondary structure. Protein Science, In Press. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
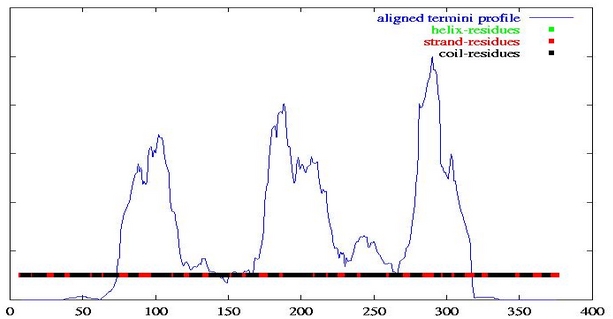
The graph is derived from the N- and C-termini positions from PSI-BLAST local alignments. In this way, large values indicate regions where sequence discontinuities occur, thus indicate putative domain boundaries. Below this is given the predicted number of domains and the positions of domain boundaries predicted from the peaks in this graph. In general, this graph should always be visually inspected to confirm the predicted number of domains and possible domain boundaries since a large degree of variation is possible, due to aspects such as disorder and variation in the domain linker region, which may not always be accurately handled by the automatic peak detector. In this case 4 domains are predicted with the domain boundaries at residue positions 102, 188, 290.
Following the PSI-BLAST derived results is the DomSSEA results. PSIPRED is used to predict the secondary structure of the query sequence, and this secondary structure is then aligned against the DSSP determined secondary structures over a complete fold library. The SSEA (Secondary Structure Element Alignment) scoring scheme is employed to carry out this matching by aligning complete secondary structure element. The reasoning behind this is that secondary structure is more conserved than sequence and thus more remote structural relationships will be detected. Once a fold is detected which matches the secondary structure of the query sequence, the domain boundaries of the matched fold are simply transferred to the query sequence. In this way, the method also predicts putative folds for the individual domains, given in terms of SCOP codes. In this case the best secondary structure match is with itself, chain A of the PDB structure 1CS6. Clearly this is a completely accurate prediction ! If the sequence was actually novel and not already in the PDB, the best match would be to chain B in PDB structure 1BIH, again predicting a 4 domain protein with boundaries at residue positions 99 ,207 and 293. In this case the domains are all predicted to be the same fold, with SCOP code b.1.1.4 (Immunoglobulin-like beta-sandwich). The third prediction is only for two domains and is a false positive. The fourth prediction is again for 4 domains with the domains having the fold given by SCOP code b.1.2.1., this is again an Immunoglobulin-like beta-sandwich, although in a different super-family (Fibronectin type III).
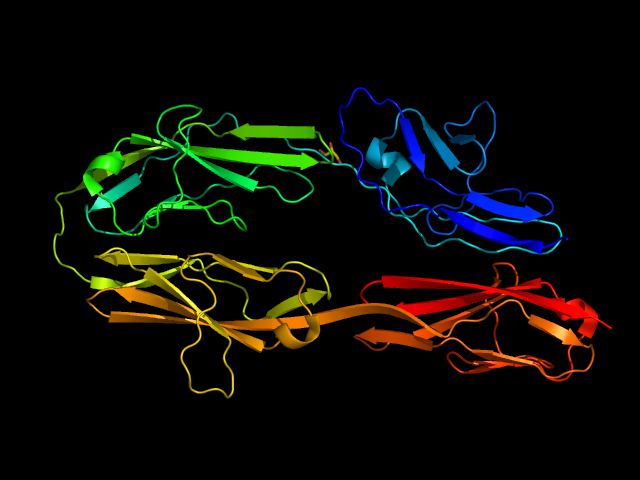
So there is a general consensus that the structure is a 4 domain structure with identical Immunoglobulin-like beta-sandwich domains. The following graphic shows that this is the correct prediction: